
Speaking the Language of Life: How AlphaFold2 and Co. Are Changing Biology | by Salvatore Raieli | Aug, 2022
AI is reshaping research in biology and opening new frontiers in therapy
Proteins are the fundamental building blocks of life, and are involved in any process in the cell. Their unique structures and diverse functions allow them to perform any task in the cell. DNA and RNA can be seen as the ROM and RAM memory.
Understanding the structure and the functions of proteins takes considerable effort from the scientific community. Last year, Alpha-fold2 revolutionized how to predict the protein structure. This week (in collaboration with the European Institute of Bioinformatics) they released the most complete database of predicted 3D structures of human proteins. This article is meant to discuss why predicting proteins is hard, why it is important, and how AI and the latest research could impact the future.
Why so much hype about proteins and their structure?
Proteins are the minuscule motors of any organism, from the unicellular to the much more complex organisms such as humans. Notwithstanding we have only 20 amino acids as building blocks of proteins, we can have infinite proteins with different shapes and functions.
Proteins are implicated in allowing the functions an organism has to perform to live, grow, and reproduce. In fact, any cell uses proteins to digest, reproduce, sustain itself, and much more. Proteins can achieve all this thanks to their unique structure, and their structure shapes their function. A newly generated protein sequence (or peptide) reaches its final shape during a process called folding.
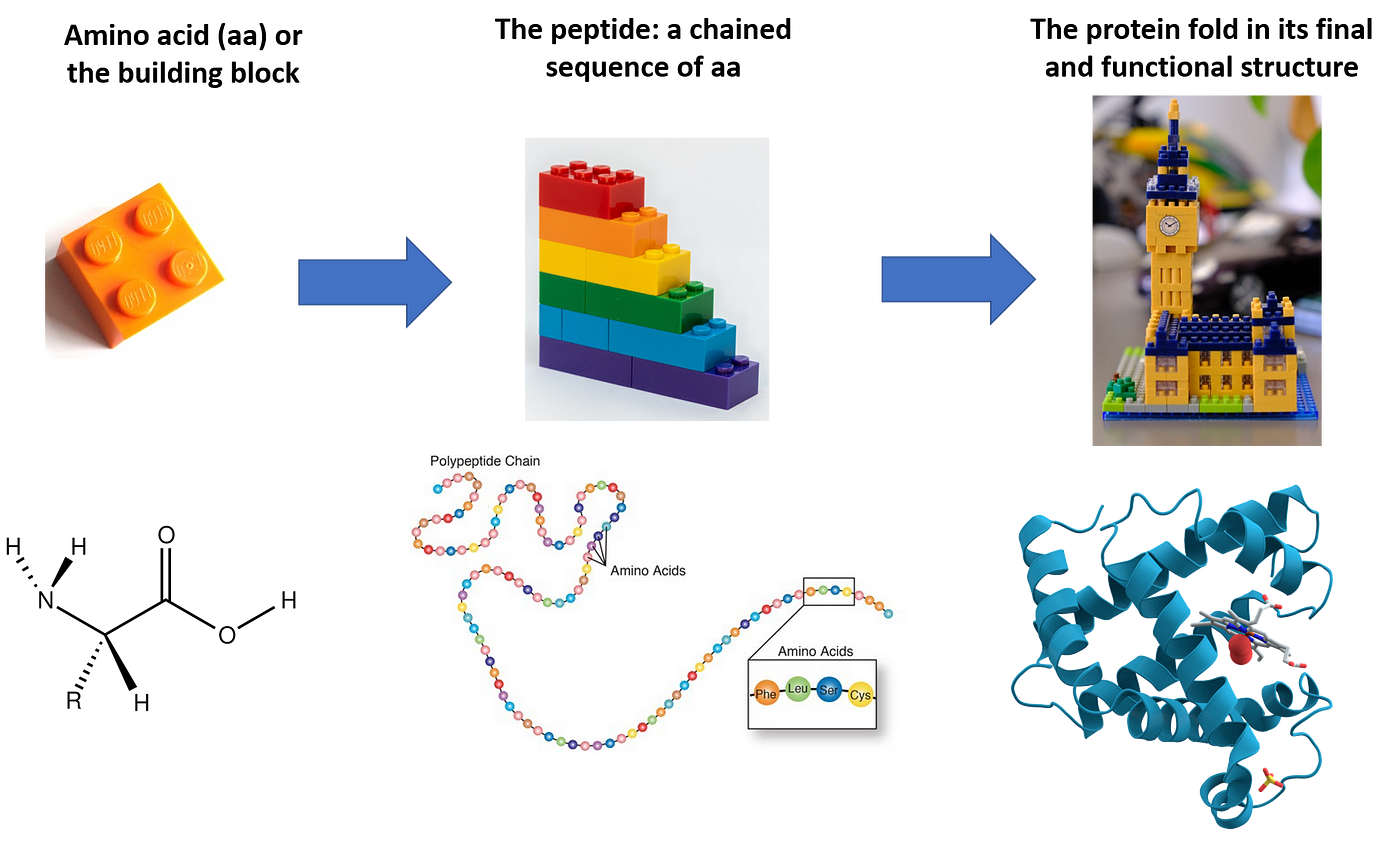
However, mutations in the DNA can change the protein sequence and lead to structural modification in the protein. The structure can be so altered the function loses its function. This is the basis for many genetic diseases or happens during cancer.
There are also cases when the protein is folded in the wrong structure and this is the basis of different diseases. For example, this is the case for Alzheimer’s disease where abnormally folded Amyloid-beta proteins are accumulated in the patient’s brain. Prion diseases are also other examples, where the pathological agent is the misfolding of a protein.
The infinite chessboard where proteins play
In the 1950s, Shannon estimated that it could be possible to play around 10**120 (10 power to 120) games. Taking into account that in the known universe there should be no more than 10**82 atoms, this is not a small number.
In 1969, Levinthal noted that since a peptide (a group of amino acids) has a large number of degrees of freedom, it could end in 10**300 possible conformations. If a protein would try all the possible combinations, it would take longer than the age of the universe. Since a protein folds (finds the correct conformation or structure) in a few milliseconds, this was called Levinthal’s paradox.
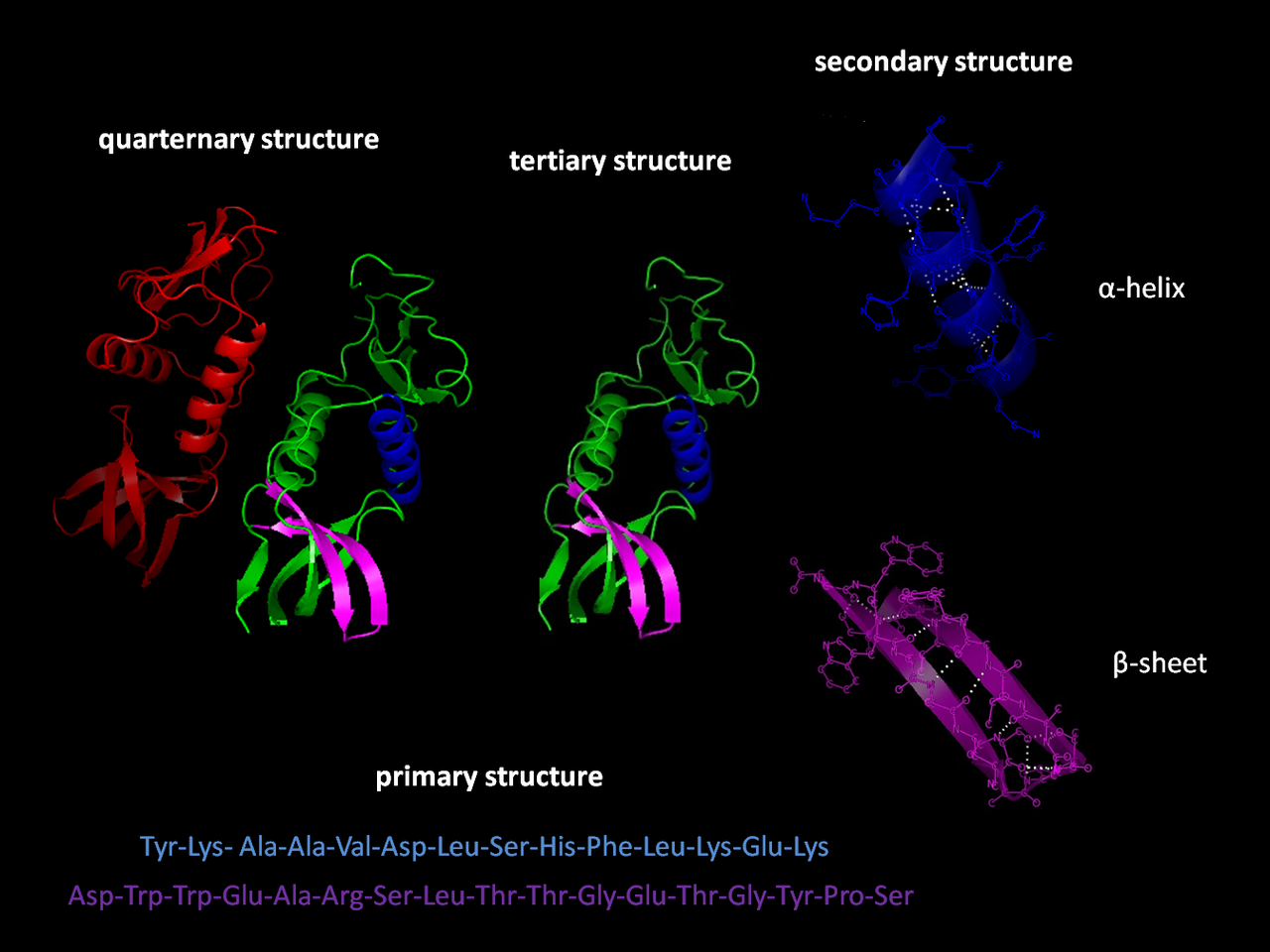
The structure of the protein defines its function and it is important for designing therapeutic drugs. Traditionally researchers have relied on experimental approaches such as X-ray crystallography or cryo-electron microscopy (cryo-EM). However, while X-ray crystallography is returning a detailed structure, it is laborious and expensive (even $100,000 for protein in some cases). Thus, many research groups tried to develop algorithms to predict the structure from the sequence.
Despite the fact that a sequence maps 1–to-1 to a 3D structure it is terribly hard to predict a structure from the sequence. A single mutation can change the structure, and two very different sequences can hand in a similar structure. Moreover, the peptide is very flexible and can rotate in multiple ways (in addition amino acids have a lateral chain that can rotate as well). In addition, we have experimentally determined around 100,000 protein structures (protein data bank) and we have millions of sequences with unknown structures.
How proteins are the languages of living organisms
Since proteins are sequences of amino acids, there is fascinating parallelism between human language and protein sequences:
- Hierarchical organization: characters are organized in words, words are organized in sentences, and sentences in paragraphs. Paragraphs can also be combined into longer texts. In the same way, the protein alphabet is composed of 20 amino acids, which can be combined in secondary structures (generally with a functional or structural role), and secondary structures in tertiary structures. Proteins can be assembled in sophisticated complexes.
- Typos and grammar: while typos in the word can alter dramatically the meaning of a sentence (a typo once cost ten million dollars to a travel agency), you can also write grammatically correct sentences that have no sense. Similarly, mutations can disrupt the function of a protein and lead to disease.
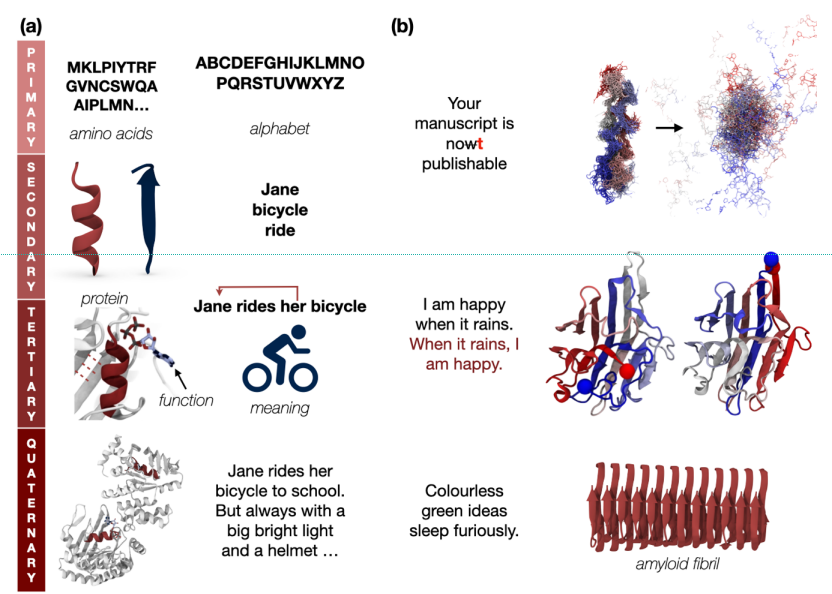
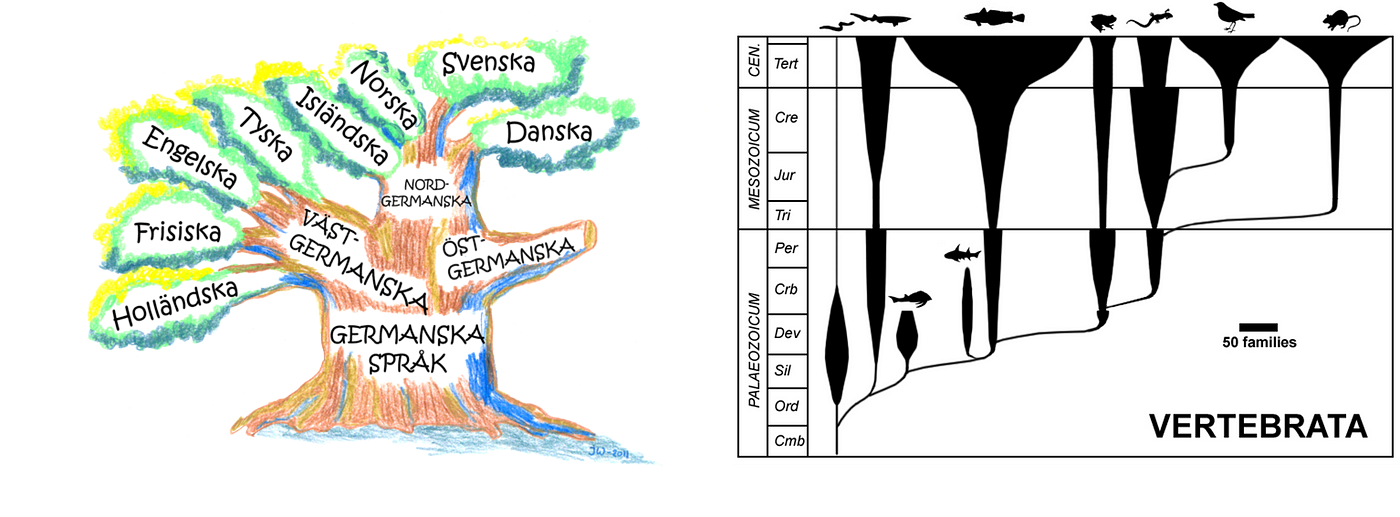
- Evolution: languages are not static; they are constantly evolving to express new concepts and reflect changes in our society. While today there are more than 8000 languages (grouped in 140 families), they probably originated from a common ancestral language that was spoken in central Africa (around 50,000–70,000 years ago). All the organisms (and their proteins) seem to be evolved from a universal ancestor around a billion years ago.
- Dependency: in the human language, words are interconnected with the surrounding words. The meaning of a word is dependent on the context. Moreover, we have long-term dependency which can be also between different sentences. There are interactions in the protein 3D structure, amino acid at the beginning of the sequence can interact with the amino acid at the end of the protein sequence. At the same time, you have interaction between secondary structures in the protein and among proteins.
However, there are some differences as well. We do not know sometimes the boundaries of words when tokenizing a protein. Moreover, there are many proteins that do not have an assigned function (we still have to decipher them).
Tackle proteins with transformers
Transformers, attention mechanisms, and derived models have had huge success in natural language processing and beyond. Initially designed to handle sequences of words have, they proved to be useful with images (vision transformers) and also with other types of data (music, graphs, and so on).
Briefly, classical transformers are constituted by an encoder and decoder and used for sequence-to-sequence problems (for example machine translation). Since their introduction they have overcome the natural language processing field, gradually substituting RNN and LSTM in almost any application. Why? Because they are much more parallelizable, they can model long dependencies, and pre-training made large transformers reusable in many fields. Most of the largest models now are transformed-based (GPT-3, BERT, and all their siblings).
Indeed, transformers have allowed AI practitioners to train models with a huge quantity of data. The amount of data is continually growing over the years (with forecasts of 80 zettabytes in 2025). The same is happening in biology and medicine: since the omics revolution we are accumulating DNA, RNA, and protein sequences. Thus, why not use transformer models to harness all this big data in biology?
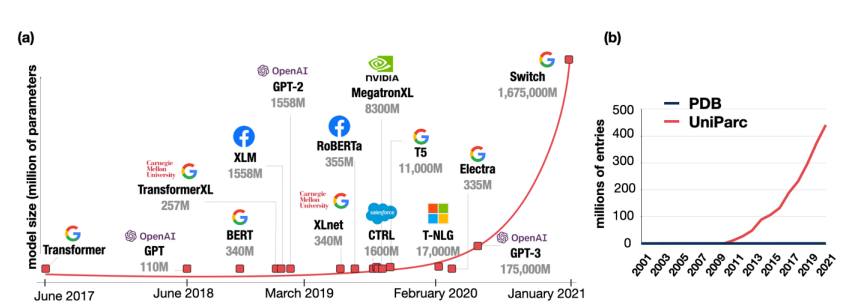
AlphaFold2 or how DeepMind surprised the whole science community
In 2020, DeepMind participated in the Critical Assessment of Structure Prediction (CASP) challenge, which is considered the most important challenge for protein structure prediction. It did not simply win, it outperformed more than 100 teams (more than 20% of gained accuracy). AlphaFold2 was capable to predict the protein structures with atomic-level accuracy. How they did do it?
The first intuition was that they could represent the data as a graph and treat the task as a graph inference problem (amino acids as nodes and the proximity as edges). Then they abandoned the convolutional network and invented Evoformer. Other interesting tricks were the use of attention mechanisms, starting from multiple sequence alignments, and end-to-end learning.
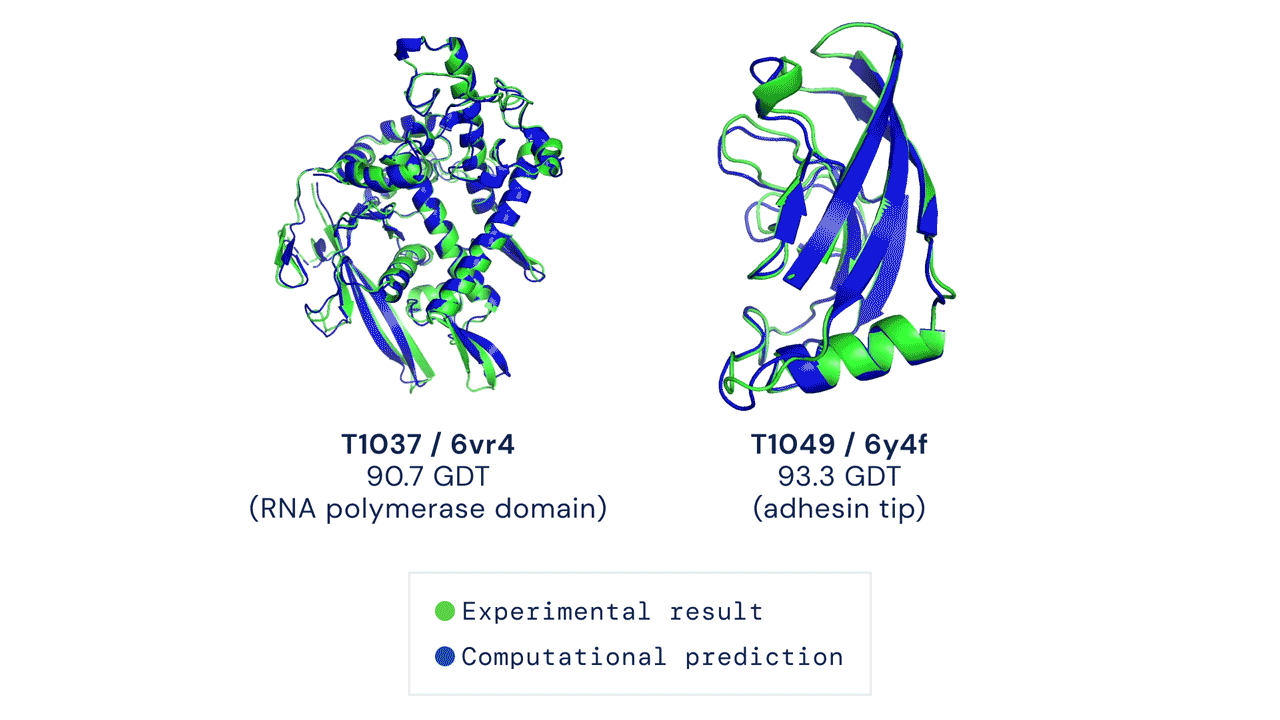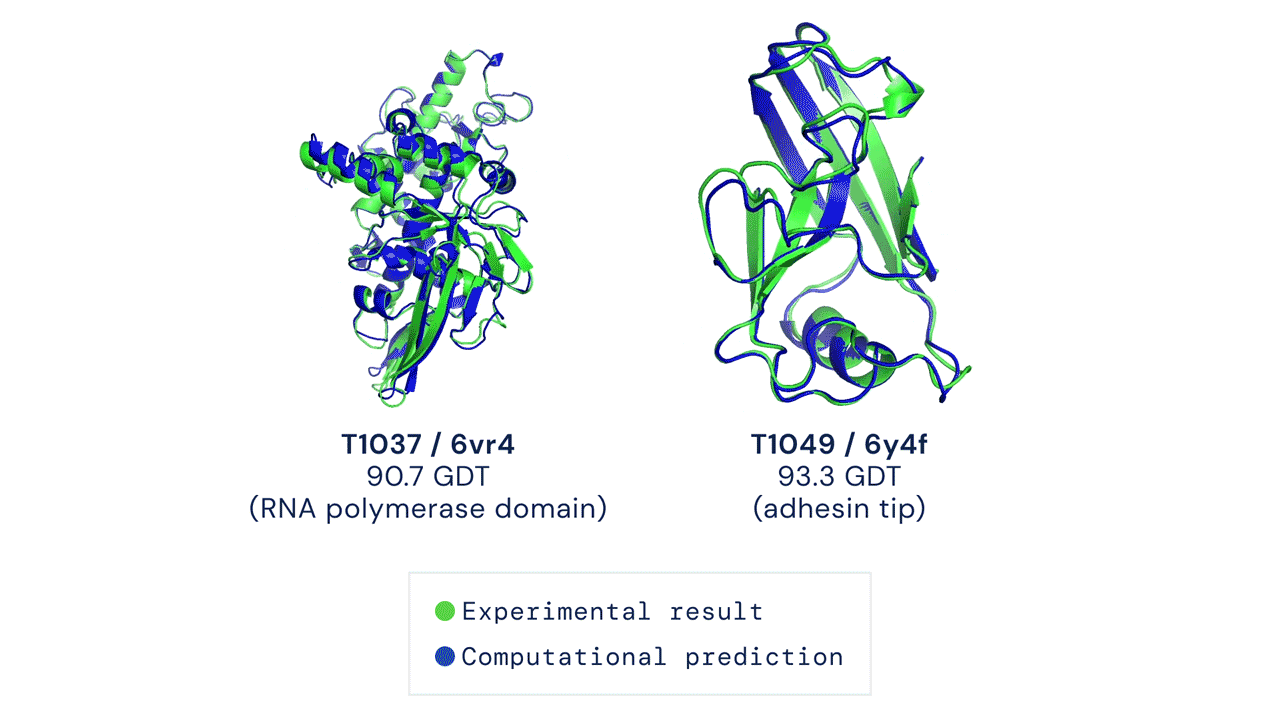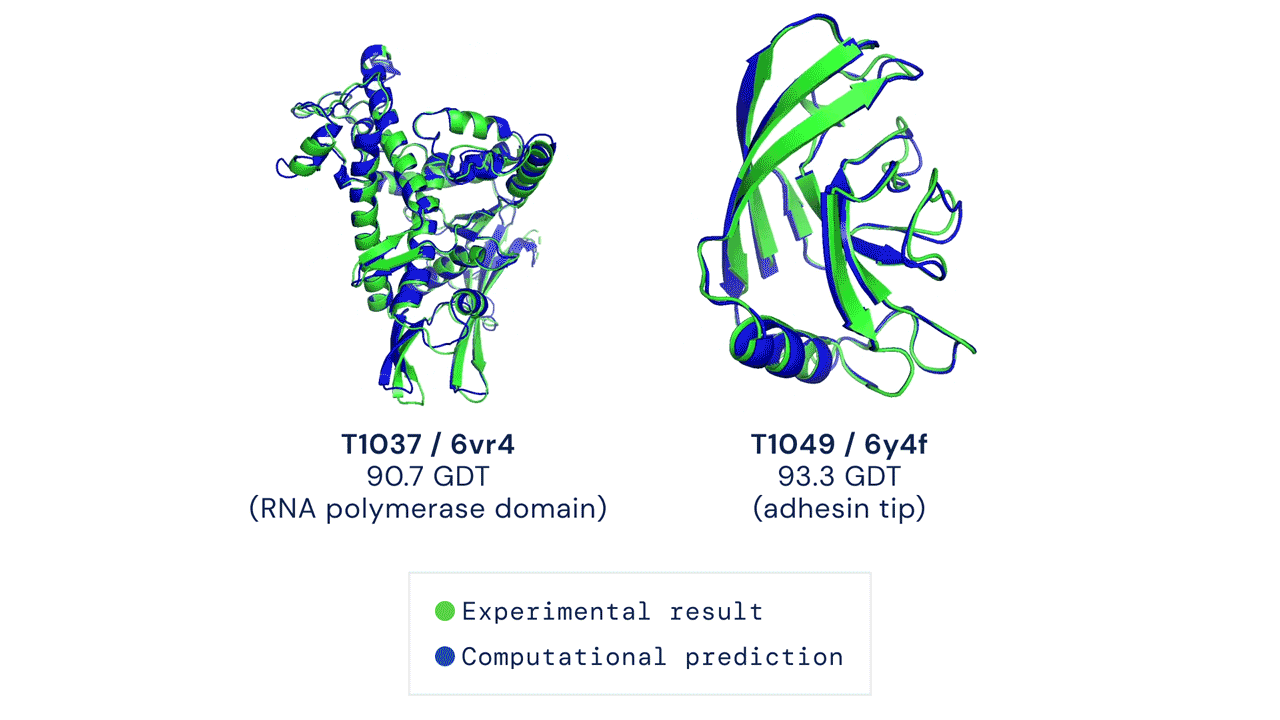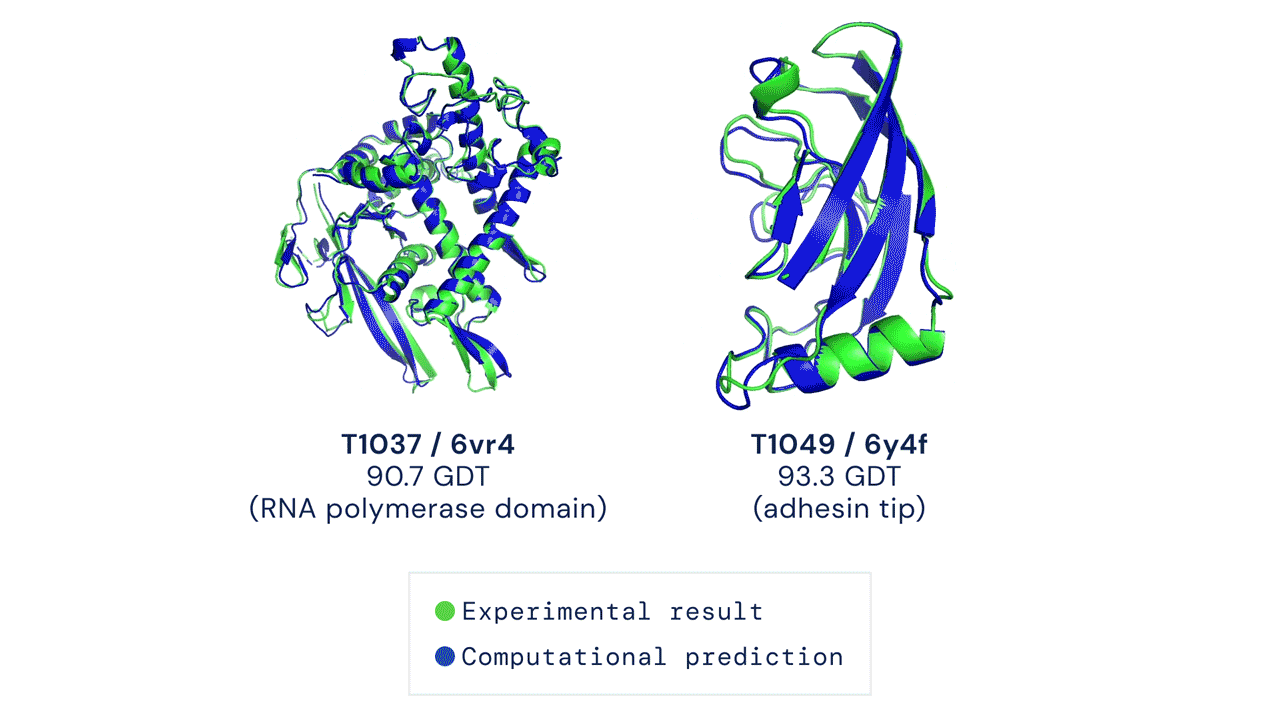
The results were impressive, even if AlphaFold had issues modeling the flexible regions or oligomeric assemblies (quaternary structures, which are even more difficult to predict). Since most of the proteins are actually working by interacting with other proteins, this is an important aspect (DeepMind released also AlphaFold-multimer, a model specifically trained for oligomer assemblies)
Back to sequence: the nemesis of AlphaFold
Recently, META presented an article at ICML presenting ESM-IF1, a model capable of doing inverse folding (predicting the sequence of a protein from its structure). Interestingly, to assemble the training set, they used AlphaFold2 to predict the structure of 12 million sequences from Uniprot. This is a clever way to overcome the limited number of protein structures that are experimentally determined.
Then, they created a model that can predict from the backbone structure (the protein structure without the amino-acids chains) the protein sequence. The amino-acid chains are important to define the function but it makes the problem more challenging and this is already a milestone. The paper described the approach of inverse folding as a sequence-to-sequence problem (using an auto-regressive encoder-decoder to retrieve the sequence from the backbone coordinates).
Interestingly, they approached the problem as a language modeling task, where they trained the model to learn the conditional distribution p(Y|X): given the spatial coordinates X, you predict the sequence Y of amino acids. Additionally, they showed that adding Gaussian noise was helping the model to train (Gaussian noise is also a new big trend).
They considered different tasks, such as predicted sequence from the backbones, multiple conformations, oligomers, and partially masked backbones. These are all possible cases in biology, as proteins can be truncated (masked parts), interact with other proteins (oligomers), or change conformations in different contexts (change in pH, presence of a medical drug). Thus, this model can be useful in different fields of research.
To understand the protein language to write our own romance
“Mother Nature has been the best bioengineer in history. Why not harness the evolutionary process to design proteins?” — Frances Arnold
First, I would like to focus on two points that have emerged until now:
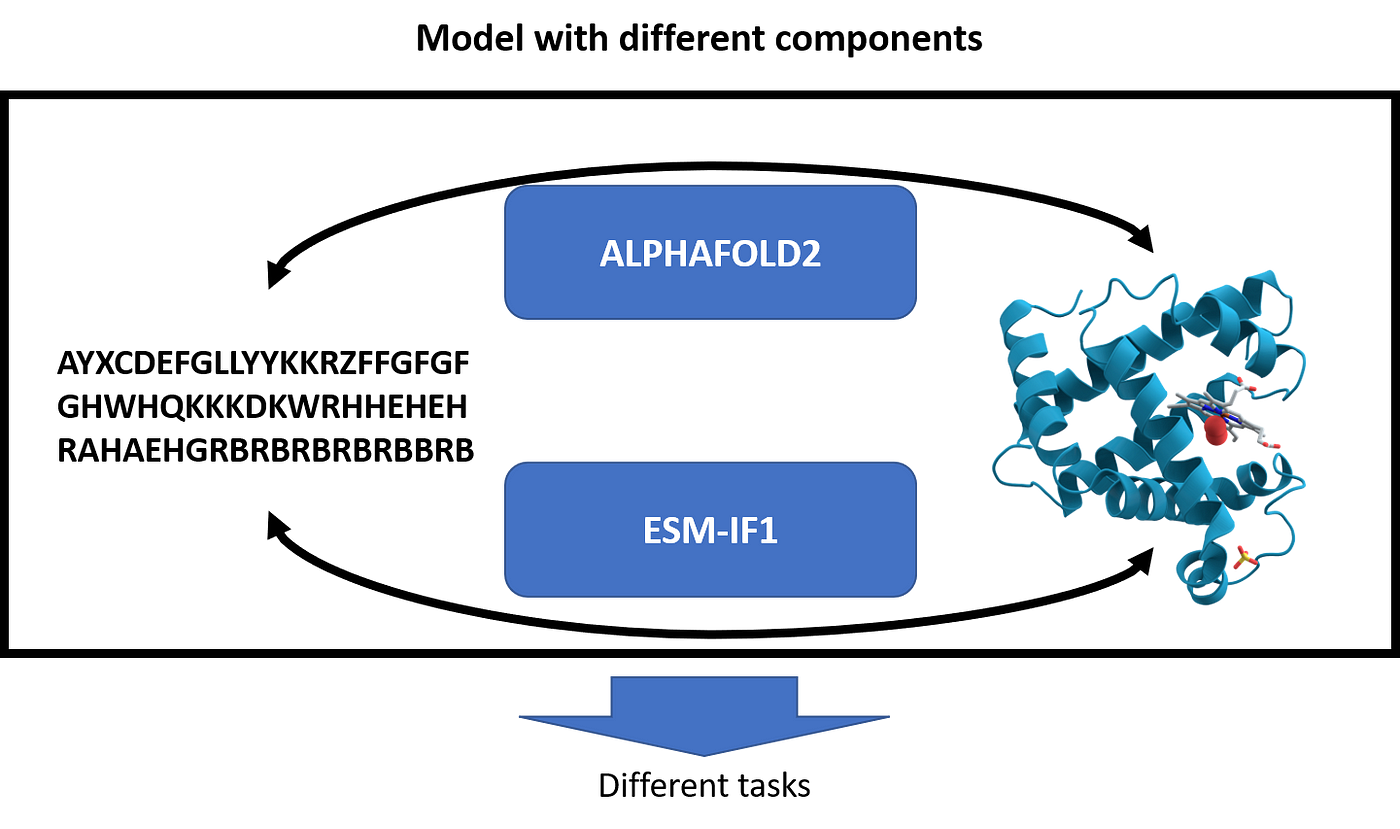
- Reformulate the task. Both AlphaFold2 and ESM-IF1 have approached the task of transforming the data in a clever way that allowed them to face the challenges in an easier way.
- AlphaFold2 as a model component. ESM-IF1 solved the lack of examples using AlphaFold2 to generate structures to train their model.
Both these ideas will influence the future, as we can easily import clever ideas from NLP (and thus use approaches that have proved to be valuable) in biological challenges. With geometric learning exploding we can also use lessons learned with graphs (but this is another story). Moreover, both AlphaFold and ESM-IF1 have been open-sourced and I am expecting they will be used in future research. In fact, AlphaFold2 is predicting the structure from the sequence while ESM-IF1 is doing the contrary, and combining them could be great for many generative tasks.
AlphaFold2 released a large dataset of predicted structures and they want to enlarge it to other organisms. Knowing the structure can be useful for many applications:
- The function of many proteins we do not know much about
- Diseases and therapeutic options. The protein structure is used to hint about the disease and you need the structure to design medical drugs that act on protein target.
- While this can be helpful to treat infective diseases, we can also design drugs against insect and plant disease
However, these models will be helpful to design new proteins for different applications. In fact, when using transfer learning the pre-trained model is not necessarily used for the original tasks. We may generate proteins with unknown function and then recover the sequence with the inverse folding. Once you have the sequence you can produce it in the laboratory the protein.
Conclusions
Ahead of us, there are exciting times. This is the dawn of more transformative research and applications. Gene editing soon enters clinics, which coupled with the idea of possibly understanding how mutation alters the studies can be the solution for many diseases (from neurological disorders to cancer). For example, Alzheimer’s and prions are diseases where the misfolding of a protein is central in the onset.
However, almost any large language model has been published by big companies. AlfaFold2 and ESM-IF1 are also the research product of two blue chip companies. Sure, these projects have created collaborations with academic institutions but since many of their applications will influence the future life of many, we also need an institutional effort.
Moreover, when we are predicting the structure for a clinical application, we also need to know why the model arrived at that prediction. Thus, we need also to discuss how explainable this technology is and how to improve its explicability.
Additional resources
- About the protein structure prediction (here) and folding problem (here, here, and here). If you are interested in the experimental side, check these videos (here, here)
- About why we need institutional efforts with large language models (here) and taming language diversity with AI (here)
- About protein de-novo design (video)
You can look for my other articles, you can also subscribe to get notified when I publish articles, and you can also connect or reach me on LinkedIn. Thanks for your support!
Here, is the link to my Github repository where I am planning to collect code, and many resources related to machine learning, artificial intelligence, and more.
AI is reshaping research in biology and opening new frontiers in therapy

Proteins are the fundamental building blocks of life, and are involved in any process in the cell. Their unique structures and diverse functions allow them to perform any task in the cell. DNA and RNA can be seen as the ROM and RAM memory.
Understanding the structure and the functions of proteins takes considerable effort from the scientific community. Last year, Alpha-fold2 revolutionized how to predict the protein structure. This week (in collaboration with the European Institute of Bioinformatics) they released the most complete database of predicted 3D structures of human proteins. This article is meant to discuss why predicting proteins is hard, why it is important, and how AI and the latest research could impact the future.
Why so much hype about proteins and their structure?
Proteins are the minuscule motors of any organism, from the unicellular to the much more complex organisms such as humans. Notwithstanding we have only 20 amino acids as building blocks of proteins, we can have infinite proteins with different shapes and functions.
Proteins are implicated in allowing the functions an organism has to perform to live, grow, and reproduce. In fact, any cell uses proteins to digest, reproduce, sustain itself, and much more. Proteins can achieve all this thanks to their unique structure, and their structure shapes their function. A newly generated protein sequence (or peptide) reaches its final shape during a process called folding.
However, mutations in the DNA can change the protein sequence and lead to structural modification in the protein. The structure can be so altered the function loses its function. This is the basis for many genetic diseases or happens during cancer.
There are also cases when the protein is folded in the wrong structure and this is the basis of different diseases. For example, this is the case for Alzheimer’s disease where abnormally folded Amyloid-beta proteins are accumulated in the patient’s brain. Prion diseases are also other examples, where the pathological agent is the misfolding of a protein.
The infinite chessboard where proteins play
In the 1950s, Shannon estimated that it could be possible to play around 10**120 (10 power to 120) games. Taking into account that in the known universe there should be no more than 10**82 atoms, this is not a small number.
In 1969, Levinthal noted that since a peptide (a group of amino acids) has a large number of degrees of freedom, it could end in 10**300 possible conformations. If a protein would try all the possible combinations, it would take longer than the age of the universe. Since a protein folds (finds the correct conformation or structure) in a few milliseconds, this was called Levinthal’s paradox.
The structure of the protein defines its function and it is important for designing therapeutic drugs. Traditionally researchers have relied on experimental approaches such as X-ray crystallography or cryo-electron microscopy (cryo-EM). However, while X-ray crystallography is returning a detailed structure, it is laborious and expensive (even $100,000 for protein in some cases). Thus, many research groups tried to develop algorithms to predict the structure from the sequence.
Despite the fact that a sequence maps 1–to-1 to a 3D structure it is terribly hard to predict a structure from the sequence. A single mutation can change the structure, and two very different sequences can hand in a similar structure. Moreover, the peptide is very flexible and can rotate in multiple ways (in addition amino acids have a lateral chain that can rotate as well). In addition, we have experimentally determined around 100,000 protein structures (protein data bank) and we have millions of sequences with unknown structures.
How proteins are the languages of living organisms
Since proteins are sequences of amino acids, there is fascinating parallelism between human language and protein sequences:
- Hierarchical organization: characters are organized in words, words are organized in sentences, and sentences in paragraphs. Paragraphs can also be combined into longer texts. In the same way, the protein alphabet is composed of 20 amino acids, which can be combined in secondary structures (generally with a functional or structural role), and secondary structures in tertiary structures. Proteins can be assembled in sophisticated complexes.
- Typos and grammar: while typos in the word can alter dramatically the meaning of a sentence (a typo once cost ten million dollars to a travel agency), you can also write grammatically correct sentences that have no sense. Similarly, mutations can disrupt the function of a protein and lead to disease.
- Evolution: languages are not static; they are constantly evolving to express new concepts and reflect changes in our society. While today there are more than 8000 languages (grouped in 140 families), they probably originated from a common ancestral language that was spoken in central Africa (around 50,000–70,000 years ago). All the organisms (and their proteins) seem to be evolved from a universal ancestor around a billion years ago.
- Dependency: in the human language, words are interconnected with the surrounding words. The meaning of a word is dependent on the context. Moreover, we have long-term dependency which can be also between different sentences. There are interactions in the protein 3D structure, amino acid at the beginning of the sequence can interact with the amino acid at the end of the protein sequence. At the same time, you have interaction between secondary structures in the protein and among proteins.
However, there are some differences as well. We do not know sometimes the boundaries of words when tokenizing a protein. Moreover, there are many proteins that do not have an assigned function (we still have to decipher them).
Tackle proteins with transformers
Transformers, attention mechanisms, and derived models have had huge success in natural language processing and beyond. Initially designed to handle sequences of words have, they proved to be useful with images (vision transformers) and also with other types of data (music, graphs, and so on).
Briefly, classical transformers are constituted by an encoder and decoder and used for sequence-to-sequence problems (for example machine translation). Since their introduction they have overcome the natural language processing field, gradually substituting RNN and LSTM in almost any application. Why? Because they are much more parallelizable, they can model long dependencies, and pre-training made large transformers reusable in many fields. Most of the largest models now are transformed-based (GPT-3, BERT, and all their siblings).
Indeed, transformers have allowed AI practitioners to train models with a huge quantity of data. The amount of data is continually growing over the years (with forecasts of 80 zettabytes in 2025). The same is happening in biology and medicine: since the omics revolution we are accumulating DNA, RNA, and protein sequences. Thus, why not use transformer models to harness all this big data in biology?
AlphaFold2 or how DeepMind surprised the whole science community
In 2020, DeepMind participated in the Critical Assessment of Structure Prediction (CASP) challenge, which is considered the most important challenge for protein structure prediction. It did not simply win, it outperformed more than 100 teams (more than 20% of gained accuracy). AlphaFold2 was capable to predict the protein structures with atomic-level accuracy. How they did do it?
The first intuition was that they could represent the data as a graph and treat the task as a graph inference problem (amino acids as nodes and the proximity as edges). Then they abandoned the convolutional network and invented Evoformer. Other interesting tricks were the use of attention mechanisms, starting from multiple sequence alignments, and end-to-end learning.
The results were impressive, even if AlphaFold had issues modeling the flexible regions or oligomeric assemblies (quaternary structures, which are even more difficult to predict). Since most of the proteins are actually working by interacting with other proteins, this is an important aspect (DeepMind released also AlphaFold-multimer, a model specifically trained for oligomer assemblies)
Back to sequence: the nemesis of AlphaFold
Recently, META presented an article at ICML presenting ESM-IF1, a model capable of doing inverse folding (predicting the sequence of a protein from its structure). Interestingly, to assemble the training set, they used AlphaFold2 to predict the structure of 12 million sequences from Uniprot. This is a clever way to overcome the limited number of protein structures that are experimentally determined.
Then, they created a model that can predict from the backbone structure (the protein structure without the amino-acids chains) the protein sequence. The amino-acid chains are important to define the function but it makes the problem more challenging and this is already a milestone. The paper described the approach of inverse folding as a sequence-to-sequence problem (using an auto-regressive encoder-decoder to retrieve the sequence from the backbone coordinates).
Interestingly, they approached the problem as a language modeling task, where they trained the model to learn the conditional distribution p(Y|X): given the spatial coordinates X, you predict the sequence Y of amino acids. Additionally, they showed that adding Gaussian noise was helping the model to train (Gaussian noise is also a new big trend).
They considered different tasks, such as predicted sequence from the backbones, multiple conformations, oligomers, and partially masked backbones. These are all possible cases in biology, as proteins can be truncated (masked parts), interact with other proteins (oligomers), or change conformations in different contexts (change in pH, presence of a medical drug). Thus, this model can be useful in different fields of research.
To understand the protein language to write our own romance
“Mother Nature has been the best bioengineer in history. Why not harness the evolutionary process to design proteins?” — Frances Arnold
First, I would like to focus on two points that have emerged until now:
- Reformulate the task. Both AlphaFold2 and ESM-IF1 have approached the task of transforming the data in a clever way that allowed them to face the challenges in an easier way.
- AlphaFold2 as a model component. ESM-IF1 solved the lack of examples using AlphaFold2 to generate structures to train their model.
Both these ideas will influence the future, as we can easily import clever ideas from NLP (and thus use approaches that have proved to be valuable) in biological challenges. With geometric learning exploding we can also use lessons learned with graphs (but this is another story). Moreover, both AlphaFold and ESM-IF1 have been open-sourced and I am expecting they will be used in future research. In fact, AlphaFold2 is predicting the structure from the sequence while ESM-IF1 is doing the contrary, and combining them could be great for many generative tasks.
AlphaFold2 released a large dataset of predicted structures and they want to enlarge it to other organisms. Knowing the structure can be useful for many applications:
- The function of many proteins we do not know much about
- Diseases and therapeutic options. The protein structure is used to hint about the disease and you need the structure to design medical drugs that act on protein target.
- While this can be helpful to treat infective diseases, we can also design drugs against insect and plant disease
However, these models will be helpful to design new proteins for different applications. In fact, when using transfer learning the pre-trained model is not necessarily used for the original tasks. We may generate proteins with unknown function and then recover the sequence with the inverse folding. Once you have the sequence you can produce it in the laboratory the protein.
Conclusions
Ahead of us, there are exciting times. This is the dawn of more transformative research and applications. Gene editing soon enters clinics, which coupled with the idea of possibly understanding how mutation alters the studies can be the solution for many diseases (from neurological disorders to cancer). For example, Alzheimer’s and prions are diseases where the misfolding of a protein is central in the onset.
However, almost any large language model has been published by big companies. AlfaFold2 and ESM-IF1 are also the research product of two blue chip companies. Sure, these projects have created collaborations with academic institutions but since many of their applications will influence the future life of many, we also need an institutional effort.
Moreover, when we are predicting the structure for a clinical application, we also need to know why the model arrived at that prediction. Thus, we need also to discuss how explainable this technology is and how to improve its explicability.
Additional resources
- About the protein structure prediction (here) and folding problem (here, here, and here). If you are interested in the experimental side, check these videos (here, here)
- About why we need institutional efforts with large language models (here) and taming language diversity with AI (here)
- About protein de-novo design (video)
You can look for my other articles, you can also subscribe to get notified when I publish articles, and you can also connect or reach me on LinkedIn. Thanks for your support!
Here, is the link to my Github repository where I am planning to collect code, and many resources related to machine learning, artificial intelligence, and more.
Denial of responsibility! Techno Blender is an automatic aggregator of the all world’s media. In each content, the hyperlink to the primary source is specified. All trademarks belong to their rightful owners, all materials to their authors. If you are the owner of the content and do not want us to publish your materials, please contact us by email – [email protected]. The content will be deleted within 24 hours.